



NMR弛豫弥散(relaxation dispersion,RD)实验是研究生物大分子动力学的一种强大工具,可以获得大分子在微秒到毫秒时间尺度上发生构象转变的结构动态信息。该技术通过探查化学交换(chemical exchange)对NMR谱线展宽的贡献来获取生物分子的激发态的信息(excited state,ES)。
要了解RD实验首先要弄明白NMR化学位移。为解释化学位移,我们以氢原子核(1H)为例。氢原子核可以看成微小磁体,它们由于核自旋角动量的量子化在NMR静态磁场B0中呈平行排列(α态)或反平行排列(β态)。其中α态处于更低的能量状态因而占略多的比重,所以整体的磁化矢量平行于B0。以一维氢谱NMR实验为例,对处于平衡态的样品(图1a)施加垂直于B0场方向的射频(radiofrequency,RF)脉冲可以使得样品宏观磁化矢量发生转动并停止在XY平面上,然后静磁场会以拉莫尔频率绕B0磁场方向进动(图1b)并产生可检测的振荡衰减的磁场信号(图1c)。对该时域信号(图1d)进行傅里叶变换即产生频域NMR谱图(图1e)。在NMR谱中,特定原子核会产生特征频率下的共振信号。该共振频率可以转换成一个与B0无关的参数,称为“化学位移”。化学位移与该原子核所处局部磁场强度成正比,而围绕原子核的电子云会在一定程度上“屏蔽”或“去屏蔽”外部磁场,因此局部磁场强度与特定的电子环境相关。这使得NMR化学位移对结构的微小变化、质子化状态和相互作用(氢键,静电相互作用等)非常敏感[1]。
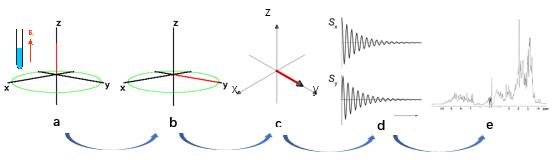
图1:化学位移的获得
要了解化学交换是如何影响NMR谱峰的,我们可以考虑某生物大分子(以RNA为例,见图2a)在两个状态之间交换:一个基态(ground state,GS)和一个激发态(excited state,ES)。我们可以将构象交换看作一个简单的二态模型(图2b),根据构象转换的正向速率和反向速率进行数值模拟。如果交换速率很慢(图2c),两种状态在测量过程中不发生构象交换,分子会清晰地分开成两群,在NMR谱上表现为两个尖锐的谱峰(图2d); 如果交换速率非常快(图2e),那么分子的“走路”和“跑步”完全无法区分,所以将以平均速率移动,在NMR谱上表现为一个尖锐的谱峰(图2f);如果交换速率是介于微秒到毫秒量级之间的中速(图2g),那么两种状态在测量过程中不断地在“走路”和“跑步”之间按照概率随机地切换,会越走越散,最后在NMR谱上表现为一个展宽的谱峰(图2h),这种现象就是弛豫弥散(RD)。
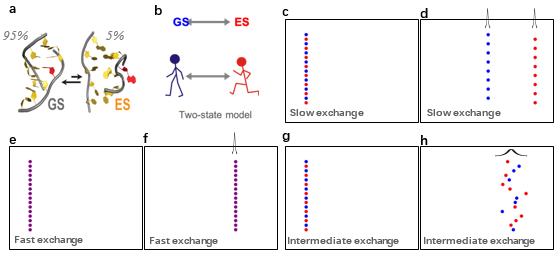
图2:化学交换对NMR谱线展宽的影响(图a引自[1])
生物大分子的激发态(ES)由于占比很低(经常在5%以下),寿命很短(微秒到毫秒的尺度),所以往往无法被直接测量到。NMR RD实验通过测量基态(GS)谱峰由于构象交换导致的展宽来间接获得难以观测的ES的结构特征以及交换过程的动力学和热力学参数。在研究生物大分子动态性方面比较常用的方法是CPMG(Carr–Purcell–Meiboom–Gill)和R1ρ。这两种实验在磁化矢量的初始状态、所使用的RF脉冲类型、测量的策略等方面都有所不同。
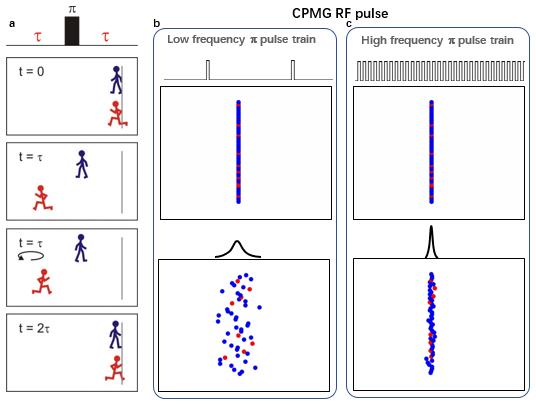
图3:CPMG实验的基本原理
CPMG实验的核心是由一组等间距的高功率180° 脉冲(又名π脉冲)组成的(图4a)。这些π脉冲可以对具有不同化学位移地核自旋演化进行重聚焦,从而抑制由化学交换造成的谱峰展宽,其原理也可以用二态模型来比较形象地解释(图3)。在GS和ES没有交换的情况下,最初GS和ES处在同一位置;经过τ时间的演化后,它们分别处于不同位置;这时对它们施加一个π脉冲会使其反向运动,那么再经过一个时间τ之后,它们将同时回到初始位置。当施加多个π脉冲组成地序列时,这些核自旋会在初始位置地左右往复运动,并且最终总能在同一时刻回到初始位置,实现重聚焦(图3a)。但是如果考虑到在这个过程中会有GS和ES的相互交换,情况就会变得不一样了。假设施加的π脉冲重复频率较低,那么每个π脉冲周期比较长,在每个周期里就有较多机会发生构象交换,结果会导致有较严重的谱峰展宽(图3b);而如果施加高频率的π脉冲,那么每个周期里就有较少机会发生构象交换,结果谱线展宽就比较小(图3c)。所以我们可以通过控制π脉冲时间间隔(τ_CP)来调节RD实验对谱线展宽的抑制作用。
假如在CPMG实验中施加的π脉冲头尾相接(τ_CP趋近于零)即直接在这个过程中施加一个持续较弱的RF脉冲就是R1r实验(图4a),我们把这个持续较弱的RF脉冲称之为“spin-lock” (SL)。在R1r实验中,由于使用的是强度较弱的脉冲,只能使较窄的共振频率范围内的原子核发生磁共振。如果我们把SL脉冲的共振频率设置成与所测量原子核的拉莫尔频率完全相同(即on-resonance,正谐共振),那么在以拉莫尔频率旋转的坐标系里观察的话B0方向的磁场被完全“抵消”,因此施加在核自旋上的有效磁场(effective field,ω_eff)就等于SL。在这种情况下的R1r实验叫做on-resonance R1r,本质上和CPMG实验是相同的,所以同样可以用CPMG的原理(图3和图4d)来理解on-resonance R1r实验。如果SL脉冲的共振频率偏离核自旋的拉莫尔频率(即off-resonance,失谐共振),那么核自旋所处的有效磁场(ω_eff)除了SL(ω_SL)外还有B0方向上的磁场失偏(offset, Ω)(图4b)。在这种情况下的R1r实验叫做off-resonance R1r 。GS和ES的Ω不同,因此它们的效应场也不同,而通过控制Ω 的总体大小或者SL的强度可以控制GS和ES有效磁场的接近程度(图4c)。此时,由于来自不同分子的核会分别以频率ωGS和ωES进行时间进动,它们将不再同步并引起相位退相干(dephasing)。这种相移会对观测到的横向弛豫速率R2有额外的GS和ES交换贡献,称为R2,obs=R2+ Rex。因为NMR线宽是与R2+ Rex成正比的,因此GS和ES的交换会表现为对NMR线宽的贡献。在R1ρ实验中Rex可以被SL强度和Ω调制(在CPMG实验则由τ_CP所调制),通过在不同SL和Ω下测定感兴趣的原子核(GS)的横向弛豫速率(R2+ Rex),可以拟合得到处于GS和ES状态下的比例分配、交换速率以及化学位移差值(图4d),从而得到本来不可见的ES的信息[1]。
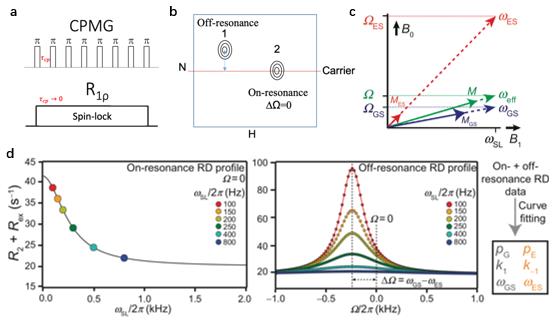
图4:R1ρ实验的基本原理(图c和图d引自[1])
有效磁场的强度决定了可测得的交换速率范围。CPMG实验中通常采用的效应场强度约为25-500 Hz,可用于测量交换速率(kex=k1+k-1)在~10s-1
化学交换过程是由于不同时间尺度上的构象转变或化学反应而发生的,通常会改变原子核所处的电子环境,从而导致部分原子核化学位移的变化。CPMG和R1ρ实验都可用于测量在微秒到毫秒时间尺度上发生的化学交换过程。在生物大分子中,这些过程往往与其功能相关,如蛋白质折叠、配体结合和催化作用[2]。通过对RD数据进行分析,可以确定构象转变或化学反应的动力学和热力学性质,并其和分子的功能联系起来。
[1] Xue, Y., Kellogg, D., Kimsey, I. J., Sathyamoorthy, B., Stein, Z. W., McBrairty, M., & Al-Hashimi, H. M. (2015). Characterizing RNA Excited States Using NMR Relaxation Dispersion. In Methods in Enzymology (1st ed., Vol. 558, pp. 39–73).
[2] Palmer, A. G., & Massi, F. (2006). Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chemical Reviews, 106(5), 1700–1719.
[3] Neudecker, P., Lundström, P., & Kay, L. E. (2009). Relaxation Dispersion NMR Spectroscopy as a Tool for Detailed Studies of Protein Folding. Biophysical Journal, 96(6), 2045–2054.
[4] Vallurupalli, P. (2009). Pramodh_Lecture Notes-Chemical Exchange, 1–33.
本文章版权归清华大学蛋白质研究技术中心核磁技术平台所有