核磁共振(Nuclear Magnetic Resonance, NMR)是研究溶液中蛋白质-蛋白质相互作用的常用方法,具有可重复性高、能提供位点特异性信息等优点,尤其是它能够可靠地测量分子间比较微弱的相互作用。其中鉴定蛋白质复合物相互作用界面运用最广泛的核磁方法是化学位移扰动(Chemical Shift Perturbation)和氢氘交换(Hydrogen-Deuterium Exchange )。但是,这两种方法都有一定的局限性,使用这些方法确定的蛋白互作界面并不总是与X射线晶体学揭示的相同。
化学位移扰动也称为化学位移映射(Chemical Shift Mapping),是通过比对配体结合前和结合后的谱峰变化来找到相互作用的界面,并定量结合的强弱。加入配体后,蛋白和配体间的相互作用会使互作界面的化学环境发生变化,导致参与互作的氨基酸残基的化学位移发生变化。我们只要找出化学位移变化明显的氨基酸残基,那么就确定了相互作用的界面。但是互作界面的改变,往往也会使位于界面附近的氨基酸残基的化学环境发生改变,导致这些氨基酸残基的化学位移变化。除此之外,蛋白质互作后,在蛋白上距离互作界面较远的区域也可能会发生构象变化(allosteric effect,别构效应),而这也会产生谱峰的变化,所以该方法有时会给我们一些错误的信息。
氢氘交换是通过检测蛋白和配体结合前后氢和氘之间交换速率的变化来确定互作界面。加入配体后,蛋白和配体会形成比较紧密的结构,相互作用区域不再暴露在水溶液中,因而H-D交换速率会变慢。但是有些结合位点本来就没有暴露在蛋白的外表面,因此在不加配体时,H-D交换速率也是很慢的。所以该方法也有明显的局限性。
这里我们介绍一种由Hideo Takahashi等人开发,基于交叉饱和(Cross-Saturation)的NMR方法[1],该方法可以更准确地识别蛋白质-蛋白质复合物中的互作界面。
交叉饱和法检测复合物互作界面
该方法的原理如图一所示,其中蛋白质I用H15均匀标记(N2H12),是我们要测量的蛋白;蛋白质II是非标记的14N1H,12C1H2和N的蛋白质I不会受到RF场的直接影响,但是,这种饱和效应可以通过交叉弛豫(Cross Relaxation)从配体分子(蛋白质II)转移到双标记的待检测分子(蛋白质I)中。如果双标记分子(蛋白质I)的质子密度足够低,则饱和转移仅限于该复合物的互作界面。我们可以通过HSQC实验检测H151-N谱峰就会有很好的效果,可以把复合物分子量的上限提升到几百kD。
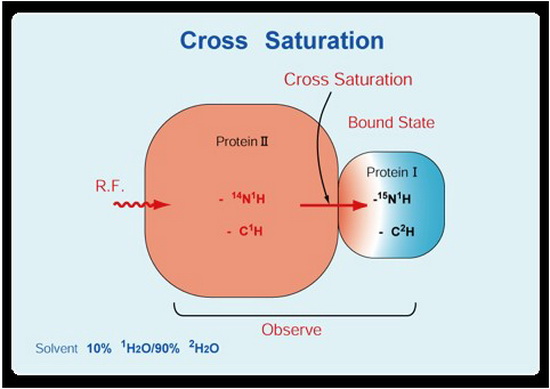
图一:交叉饱和方法原理示意图。
小贴士1:饱和原理
弛豫(Relaxation)是自旋体系受扰动后恢复到平衡态的过程。平衡态(Equilibrium)是这样一种状态,即两个能级上的大量核自旋遵从玻尔兹曼分布,且没有横向磁化矢量,也就是说系统中不存在相干性(如图二a所示)。平衡态的宏观磁化矢量(Mz)是由α和β自旋态两个能级的不均等分布引起的: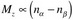
接下来我们使用弱的RF场持续照射目标自旋系统。选择的磁场要足够弱,这就意味着它的选择性足够强,以至于只有共振频率在一定范围内的自旋才会受到影响。这样照射最终会使得目标自旋在两个能级上的分布数量达到相等,从而使得宏观磁化矢量为零(如图二b所示)。在波谱学中将这种情况称为饱和(Saturation)。
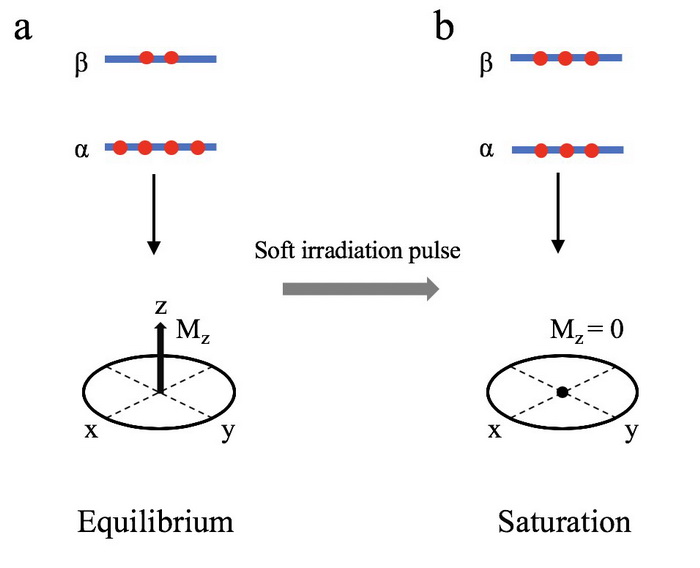
图二:平衡状态(a)和饱和状态(b)两个能级分布及其宏观磁化矢量
小贴士2:交叉弛豫原理
在双自旋系统中(如图三所示),I自旋上的极化状态(即自旋粒子在两个能级上的分布情况)随时间变化的方式不仅取决于该自旋偏离平衡态的程度,也取决于在它附近的S自旋所处的极化状态——即一个核自旋的极化状态可以通过空间上的偶极-偶极相互作用来影响它附近的另一个核自旋的极化状态随时间的变化,这种现象称为交叉弛豫(Cross relaxation)。I自旋的极化状态随时间的变化率与两个自旋间距离(r)的6次方成反比,因此随着距离的增加会迅速下降。在实践中,我们只能观察到彼此相当接近的自旋之间的交叉弛豫效应。对于质子对来说,这一距离要小于5Å才可以观察到交叉弛豫。因此,如果我们看到两个自旋之间有交叉弛豫的现象,则可以确定它们在空间上相当接近。交叉弛豫会导致我们熟知的NOE效应(nuclear Overhauser effect)。
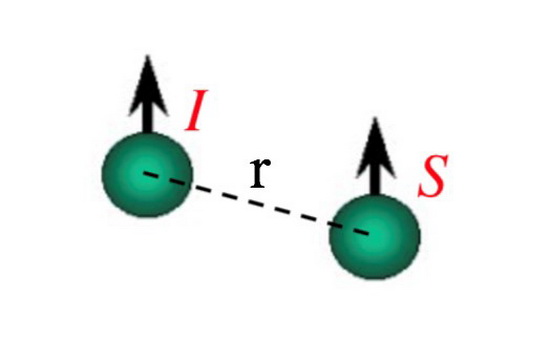
图三:双自旋系统示意图
应用交叉饱和方法检测FB-Fc复合物的互作界面
下面将介绍该方法的一个应用实例:通过交叉饱和方法检测FB–Fc复合物的互作界面。Fc为免疫球蛋白G8(IgG8)的片段,FB是蛋白A(Ig结合蛋白的一种)特异性结合Fc的部分。其中,FB是待检测蛋白,为H151-N TROSY-HSQC谱图。在图四b可以清楚地观察到饱和照射(照射时间为1.2s)会显著降低FB中一些峰的强度,这表明复合物中Fc片段的饱和效应通过互作界面转移到FB了。图四c显示,在不同的饱和照射时间(0.3、0.6、1.2和1.8s)下,受照射的NH谱峰和未受照射的NH谱峰的强度比值。从图中可以看出氨基酸N29,R28,Q11随着照射时间的增加,峰强度比值明显下降最后达到平衡,而氨基酸K59和A55在照射下,峰强度比值没有明显的下降。因此,N29,R28和Q11可以确定为参与互作的氨基酸残基。
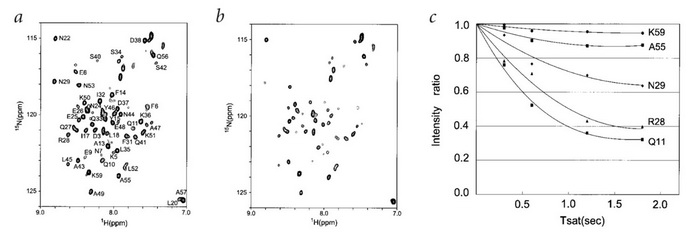
图四:无照射(a)和有照射(b)情况下,FB-Fc复合物1H-15N TROSY-HSQC谱图。(c)为饱和时间对受照射和无照射酰胺基交叉峰强度比的影响。所用的饱和时间为0.3、0.6、1.2和1.8s。
比较不同方法对FB-Fc复合物的互作界面的确定
图五abcd分别为X射线晶体学,化学位移扰动,H-D交换,交叉饱和这五种方法所得到的结果,该图所示蛋白为FB,红色显示的氨基酸残基为各方法确定的互作界面。很明显我们可以发现使用交叉饱和的方法与X射线晶体学的结果更为接近。因此,与之前使用的其他NMR方法相比,交叉饱和的方法能够更精确地确定FB-Fc复合物的界面。
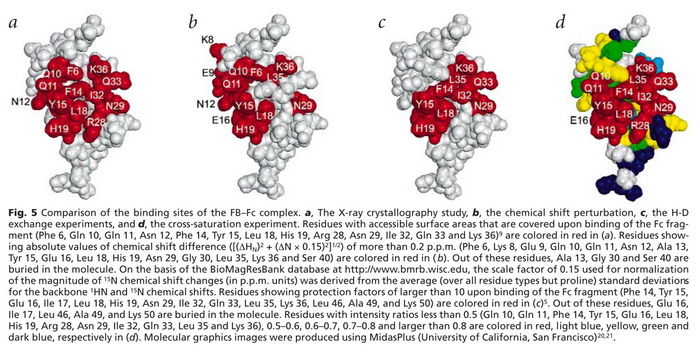
图五:不同方法确定的FB-Fc复合物结合位点的比较。a,X射线晶体学研究,b,化学位移扰动,c,H-D交换实验,d,交叉饱和法。红色显示的氨基酸残基为各方法确定的互作界面。
在鉴定蛋白质-蛋白质复合物的界面时,交叉饱和方法明显优于化学位移扰动或者H-D交换这样的传统NMR方法。这是因为该方法提取的是两个分子在空间中相互作用的直接信息。当然,该方法需要制备氘带样品,成本相对较高。期待之后科学家会开发出准确度高并且成本较低的确定复合物互作界面的核磁方法。
参考文献:[1] Takahashi, Hideo, et al. "A novel NMR method for determining the interfaces of large protein-protein complexes." Nature Structural & Molecular Biology7.3 (2000): 220-223.
本文章版权归清华大学蛋白质研究技术中心核磁技术平台所有
