作者:陈宇凌
蛋白质化学与组学平台作为校级服务平台之一,能够提供多项常规的基于质谱的蛋白质相关的技术支持,同时也在不断引进并开发新技术和新方法。为了让老师和同学们更多地了解蛋白质化学与组学平台提供的样品分析项目,我们将对平台的服务项目、分析原理和流程,以及样品要求做一个简单介绍。
表1 蛋白质化学与组学平台服务项目
一、常规服务
1. 简单蛋白质鉴定、蛋白N/C端鉴定简单蛋白质鉴定的样品一般有两种,(1)对已知序列的蛋白进行质谱验证或鉴定翻译后修饰,我们希望在样品分析前获得蛋白序列,从而更好地选择合适的蛋白酶进行酶切;如果有预测的蛋白修饰位点,也可以提前告知平台,方便我们在后续数据处理时重点确认这些被修饰的位点。(2)对未知序列的蛋白进行鉴定或检测样品中有什么蛋白及翻译后修饰的类型和位点等,我们需要知道蛋白样品的来源(种属等),以选择合适的数据库进行搜库和序列比对。蛋白N/C端鉴定主要是鉴定蛋白降解或者使用蛋白酶处理后的产物的N/C端,可以通过Edman降解法或质谱法确定蛋白的N-端。样品要求:SDS-PAGE分离的蛋白,考染或者银染(显色时间<2min )能够看到明显条带;利用质谱鉴定蛋白修饰或n/c端,需要较大的样品量(胶上有较粗的条带),以增加检测到的蛋白的序列覆盖率。蛋白溶液样品,尽量避免任何去垢剂和聚合物,因为这两种试剂会在色谱分离和质谱检测上干扰肽段的分析;同时避免样品中甘油浓度过高。对于利用edman降解法鉴定蛋白n端,样品可以是蛋白溶液或pvdf膜,如为蛋白溶液,要求蛋白浓度在1mg/ml以上,避免铵盐、甘油和去垢剂;如为pvdf膜,要求考马斯亮蓝染色后条带清晰可见。
2. 蛋白分子量的精确测定蛋白分子量的精确测定可通过MALDI质谱或蛋白质原位质谱分析实现。MALDI适合检测较小分子量的蛋白,对于较大蛋白或蛋白复合物,更多地使用蛋白质原位质谱。蛋白质原位质谱分析,是在不酶解前处理的情况下,通过质谱仪直接测定蛋白质或蛋白质复合物的质量,研究蛋白质修饰或蛋白质和配体的相互作用,包括蛋白质-蛋白质、蛋白质-小分子的相互作用,提供蛋白复合物的组成比例,步骤如图1所示。样品要求:蛋白质原位质谱分析对样品纯度要求较高,需要通过凝胶色谱柱或超滤离心管去除干扰的小分子纯化蛋白或蛋白复合物;尽量使用挥发性的盐溶液作为缓冲液,如0.1M的醋酸铵,pH值在6~8之间。仪器的进样浓度为1~20μM,进样体积为1~2μl,尽可能提供高浓度的样品,方便进行测试条件的优化。
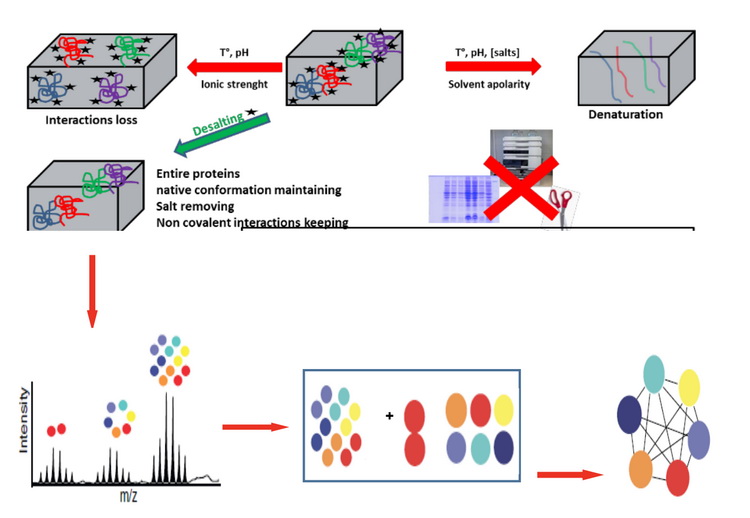
图1 蛋白质原位质谱分析步骤
3. 复杂样品的相对定量蛋白质分析
3.1 基于SILAC标记的相对定量蛋白质组学分析细胞培养中氨基酸稳定同位素标记(SILAC, Stable Isotope Labeling with Amino acids in Cell culture),是2002年Mann实验室发展的一种定量蛋白质组学技术。SILAC定量蛋白质组学分析需要在细胞培养时掺入同位素标记的赖氨酸和精氨酸,常用的赖氨酸的同位素标记为4,4,5,5-D4、13C6、13C615N2;精氨酸的同位素标记为4,4,5,5-D4、13C6、13C615N4;通过这两种氨基酸不同同位素标记状态的搭配,SILAC定量最多可以实现三组样品之间的比较,赖氨酸和精氨酸的搭配分别为K0R0(轻标)、K4R6、K8R10,如图2所示。基于SILAC定量蛋白质组学分析不仅可以提供不同条件(如基因编辑、药物刺激等)下细胞、线虫或小鼠的蛋白质组学的变化,还能对蛋白合成速率进行定量分析,将正常培养的细胞的培养基更换成稳定同位素氨基酸的培养基,新合成的蛋白和已有的蛋白分别为重标氨基酸和轻标氨基酸标记的蛋白,通过测定各时间点的各蛋白质的重轻比,得到每种蛋白质的合成速率。此外,SILAC标记的定量蛋白质组学还常被用于免疫沉淀中互作蛋白的鉴定。
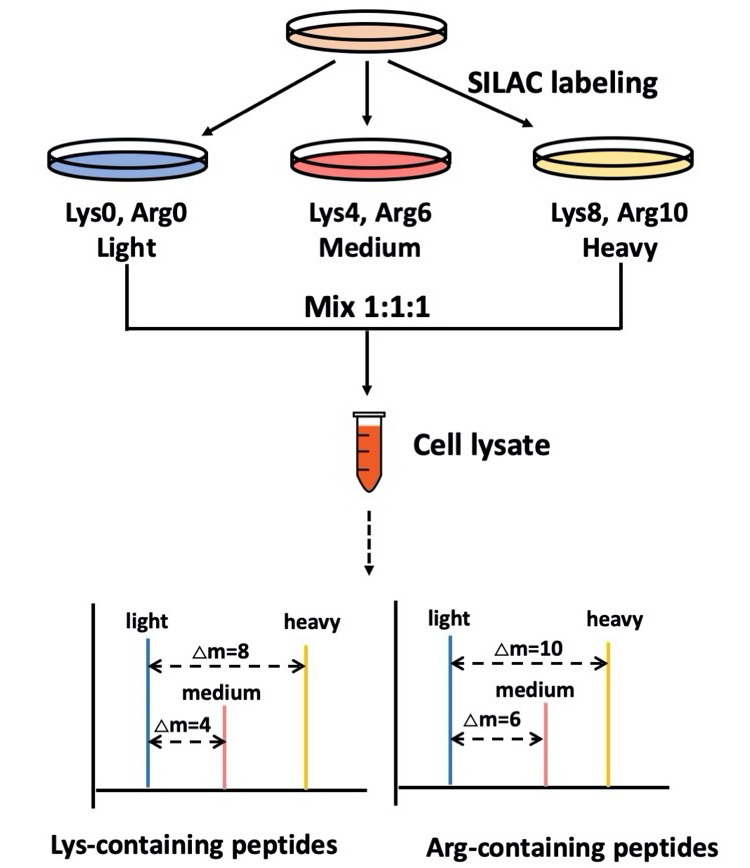
图2 基于SILAC定量蛋白质组学技术流程
样品要求:平台接收SILAC试剂标记的蛋白溶液或蛋白胶,在正式实验前,细胞应在含稳重同位素氨基酸的培养基中培养五代以上,提取蛋白跑胶,利用质谱检测蛋白中稳重同位素氨基酸的掺入率,以保证含稳重同位素的赖氨酸和精氨酸在蛋白中的掺入率达到95%以上。如为蛋白溶液,细胞裂解应使用含8 M尿素和蛋白酶抑制剂(protease inhibitor cocktail)的PBS(pH7.4~8.5),尽量保证蛋白浓度在1mg/ml以上,将不同标记的样品等量混合,提供100微克总蛋白即可;如为SDS-PAGE,对细胞裂解液没有要求,不同标记的样品等量混合后跑胶分离,蛋白胶上有较为清晰的条带,蛋白总量越高,检测到的蛋白数目越多。
3.2 基于TMT标记的相对定量蛋白质组学分析TMT(Tandem Mass Tag)是由美国Thermo Scientific公司研发的一种多肽体外标记技术,现有TMT2plex、TMT6plex和TMT10plex,可以分别实现最多2组样品、6组样品和10组样品之间的相对定量分析。TMT试剂由一个与伯铵反应的琥珀酰亚胺酯、一个中间连接臂、一个二级质谱报告离子组成,可以和肽段N端以及赖氨酸侧链的伯铵基团反应,标记不同TMT分子的肽段在一级图上分子量完全相同,经过HCD(High collision dissociation)碎裂后,在二级质谱图上实现同一个肽段在不同样品中的相对定量,分析流程如图3所示。相比起SILAC,基于TMT标记相对定量蛋白质组学适用范围非常广,可以对细胞、组织、血清、血浆、尿液或其他各种类型的蛋白组进行分析,并且可以同时做到最多十组样品的相对定量。样品要求:最好是蛋白溶液样品,细胞裂解使用含8 M尿素、蛋白酶抑制剂(protease inhibitor cocktail)的PBS,pH7.4~8.5,测定完蛋白浓度,保证蛋白浓度在1mg/ml以上,每个样品100微克蛋白即可。具体可参考平台提供的《基于TMT标记相对定量蛋白质组学实验步骤》。
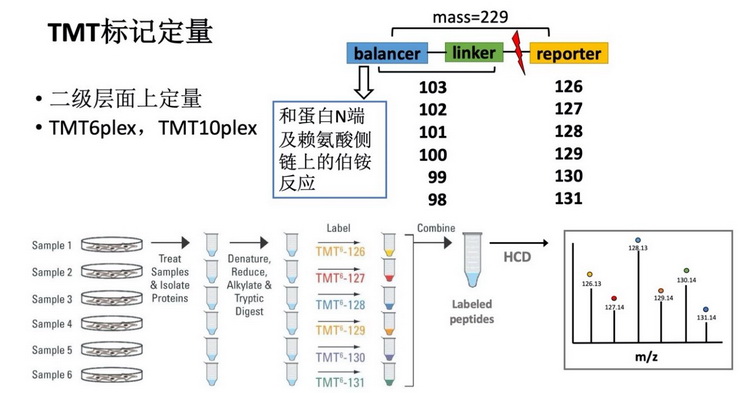
图3 基于TMT相对定量蛋白质组学分析技术流程(以TMT6ple
x为例)
3.3 非标记相对定量蛋白质组学分析非标记相对定量蛋白质组学分析一般是基于肽段在质谱上的响应强度,对不同样品中的同个蛋白进行相对定量分析。该定量方法样品前处理步骤比较简单,可实现对多个样品进行相对定量分析,但受样品前处理和仪器状态的影响比较大,进行比较的样品需要在同一台仪器相同状态时进行分析,结果重复性和平行性不如同位素标记的定量分析。非标记相对定量蛋白质组学常被用于免疫沉淀中互作蛋白的定量分析,也可用于组织、血液、细胞等样品中蛋白组的相对定量分析。样品要求:如为蛋白溶液,细胞裂解使用含8 M尿素、蛋白酶抑制剂(protease inhibitor cocktail)的PBS,pH7.4~8.5,尽量保证蛋白浓度在1mg/ml以上,100微克蛋白即可;如为1D SDS-PAGE,对细胞裂解液没有要求,蛋白胶上有较为清楚的条带,蛋白总量越高,检测到的蛋白种类越多。互作蛋白的鉴定一般使用蛋白胶,可去除实验过程中引入的去垢剂等,也可以使用柱上酶切的方式,但后者需要在酶切前用缓冲液(如PBS、NH4HCO3等)反复洗填料,去除填料上的去垢剂。
4. 复杂样品翻译后修饰谱的蛋白质组学分析
蛋白质组学和化学平台发展和优化了磷酸化修饰谱、乙酰化修饰谱、泛素化修饰谱和谷胱甘肽化修饰谱的蛋白质组学分析方法。其中,磷酸化修饰谱的鉴定使用二氧化钛富集磷酸化修饰肽段,一次实验可鉴定一万五千以上的磷酸化修饰位点;乙酰化修饰谱的分析使用乙酰化抗体来富集乙酰化肽段,可一次鉴定到四千以上的乙酰化修饰位点;泛素化修饰谱使用抗GG-K的抗体富集带GG-K的肽段(胰酶水解泛素化修饰的蛋白产生赖氨酸上留下GG的肽段),可一次鉴定五千以上的泛素化修饰位点。样品要求:蛋白翻译后修饰谱的分析需要在蛋白提取时,不仅添加常规的蛋白酶抑制剂,还需添加相应的去修饰蛋白酶抑制剂,具体请参考平台提供的翻译后修饰谱分析的实验步骤。所有翻译后修饰谱分析最好利用蛋白溶液,因为修饰蛋白的比例比较低,磷酸化修饰谱的分析要求蛋白量>2mg,乙酰化和泛素化修饰谱的分析要求蛋白量>20mg。
二、基于质谱的微量样品、蛋白质构象变化和靶向蛋白的分析
1. 检测蛋白构象变化的氢氘交换质谱分析
蛋白质氢氘交换质谱(HDX MS,hydrogen deuterium exchange mass spectrometry)是一种研究蛋白质空间构象的质谱技术,其原理是蛋白在重水溶液中,可发生蛋白的氢原子与重水的氘原子的交换反应,蛋白表面酰胺键的氢比蛋白内部或参与蛋白-配体相互作用酰胺键的氢交换速率快,通过质谱检测确定蛋白质不同序列片段的氢氘交换速率,从而构建蛋白空间结构变化的信息,步骤如图4所示。蛋白质氢氘交换质谱可用于分析蛋白质在溶液中的折叠状态,聚合状态和构象变化;也用于检测蛋白质与其他分子间的相互作用与结合位点,如蛋白与蛋白、蛋白质与多肽、蛋白质与活性小分子、蛋白质与核酸、蛋白质与细胞膜等之间的相互作用。样品要求:蛋白-配体和蛋白样品,样品浓度>1μg/μl,需要10-20μl,避免去垢剂。平台提供蛋白质氢氘交换质谱分析需要用到的水溶液和重水(D2O)溶液。
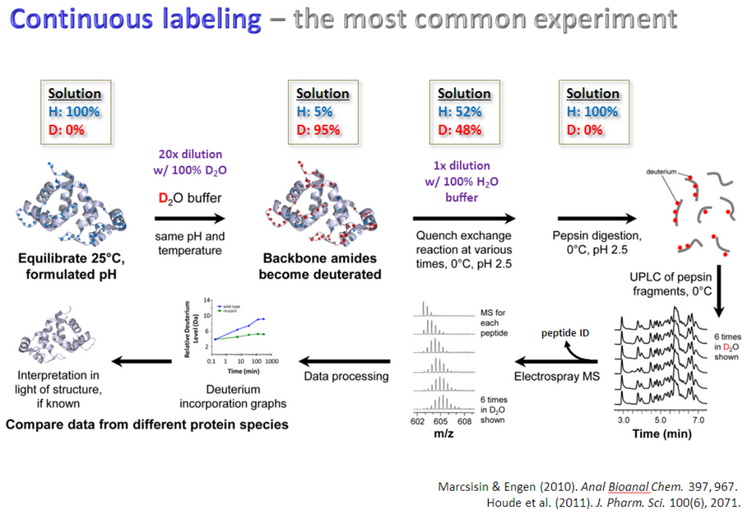
图4 蛋白质氢氘交换质谱分析步骤
2. 微量样品蛋白质组学分析
在一个细胞中,蛋白的拷贝数分布范围较广(10~108),平均拷贝数为50000个,质谱的检测灵敏度约为1-10fmol,平均需要十万个细胞才能检测到八千种蛋白。对于较少细胞量或组织量的样品,我们发展了微量样品蛋白质组学分析技术,通过结合长分析柱和长梯度分离,可在数千个细胞中鉴定到超过6000种不同的蛋白并对其进行定性定量分析。
3. 基于质谱的Western BlottingWestern Blotting是实验室中最常用的技术之一,用来检测靶标蛋白的表达量。由于抗体应用的局限性,我们提供基于质谱技术的Western Blotting,可比较不同样品中靶标蛋白或其翻译后修饰水平的变化,甚至可以检测靶蛋白的绝对表达量,如图5所示。方法一:使用普通相对定量质谱,检测靶蛋白或其翻译后修饰在不同样品中的表达量变化。方法二:合成靶蛋白的1~2条肽段,结合TMT标记技术和DDA检测模式,检测靶蛋白或其翻译后修饰在不同样品中的水平。方法三:合成靶蛋白的1~2条肽段,结合质谱的PRM采集模式检测靶蛋白或其翻译后修饰在不同样品中的相对水平。方法四:和方法二类似,采集模式换成PRM,只检测对应出峰时间的特定肽段。

图5 蛋白靶向鉴定
从左到右依次为方法二、方法三、方法四的实验流程
基于高分辨率质谱的PRM技术是利用四级杆质量分析器的离子选择功能,选择性地检测目标肽段离子,随后在HCD碰撞池中将该离子碎裂,检测所有的碎片离子。方法一适用于高中等丰度的靶蛋白的检测,方法二、三、四适用于丰度低的靶蛋白或翻译后修饰的检测。
4. 蛋白绝对定量分析
不同蛋白因为大小、氨基酸组成等不同,无法用肽段离子的峰面积或信号强度来确定其蛋白在一个样品中的丰度,需要利用iBAQ、emPAI、APEI等蛋白质组学方法,目前应用比较广泛的是iBAQ技术。对于少数目标蛋白的绝对定量分析,只需要分别合成目标蛋白对应的肽段或稳重同位素标记的肽段,进行质谱检测。对于蛋白质组的绝对定量分析,需要用到Sigma的UPS2标品, UPS2由48个人源蛋白组成,每个蛋白的量在50pmol~500amol之间,分析步骤如图6所示。
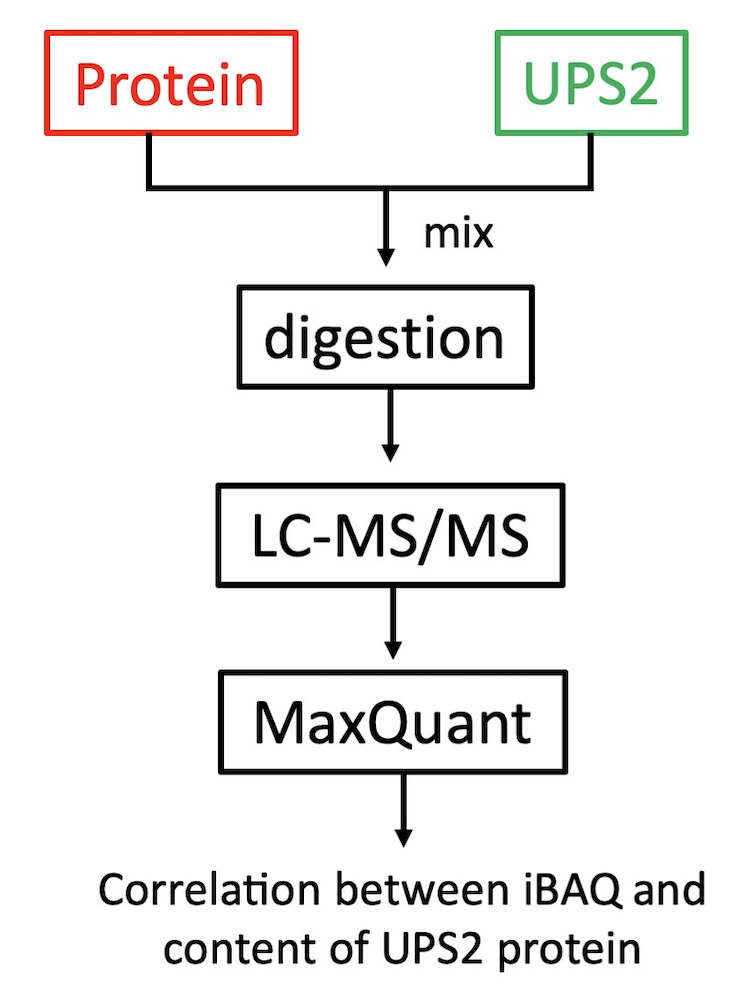
图6 蛋白绝对定量分析
5. 抗体全序列测定
平台提供单克隆抗体的全序列精确测定,需要50微克以上的单克隆抗体蛋白。
6. 组蛋白翻译后修饰定性及定量分析
组蛋白的翻译后修饰是表观遗传的重要调控途径之一,但由于组蛋白的序列中富含赖氨酸和精氨酸,以及组蛋白密码的复杂性,导致质谱鉴定组蛋白上的修饰类型和位点非常困难。为解决这个问题,我们建立了通过PPA-NHS封闭赖氨酸氨基的分析策略,并使用Epiprofile软件(Dr. Zuofei Yuan)对组蛋白的翻译后修饰情况进行定性定量分析,如图7所示。这一分析需要提供大于5×106以上的细胞量。

图7 组蛋白翻译后修饰定性及定量分析
7. 细胞膜蛋白组学分析利用不穿透细胞质膜的Sulfo-NHS-S-S-Biotin生物素化试剂与细胞膜表面蛋白的伯胺(-NH2)反应,从而标记细胞膜表面的蛋白,之后利用Streptavidin琼脂糖凝胶珠富集被标记的膜表面蛋白,随后将富集到的蛋白进行常规胶内酶解、质谱鉴定。步骤如图8所示,本实验需要107以上个细胞。
8.鉴定活性分子靶标蛋白的热蛋白组分析
热蛋白质组学分析(thermal proteome profiling,TPP),是基于蛋白和活性分子的结合可以稳定蛋白,减少蛋白随温度升高产生的聚沉这一原理,如图9所示,发展出来的用于鉴定活性分子靶标蛋白的蛋白质组学方法。该方法通过测定加药前后,不同加热温度下蛋白在蛋白溶液中的相对含量,绘制蛋白随温度变化的溶解曲线,计算蛋白聚沉50%时的温度(melting temperature,Tm),Tm在加药后发生变化的蛋白被认为是活性分子的潜在靶标蛋白。固定药物浓度的TPP实验步骤如图10所示,需要细胞量为5×106。除此之外,还可以通过不同的药物浓度梯度和温度梯度的结合,鉴定每个潜在的靶标蛋白对药物分子的亲和力。
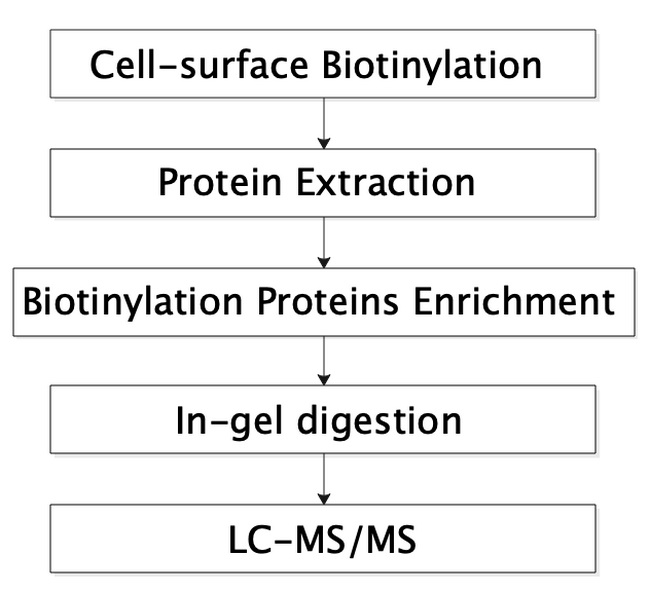
图8 细胞膜表面组学
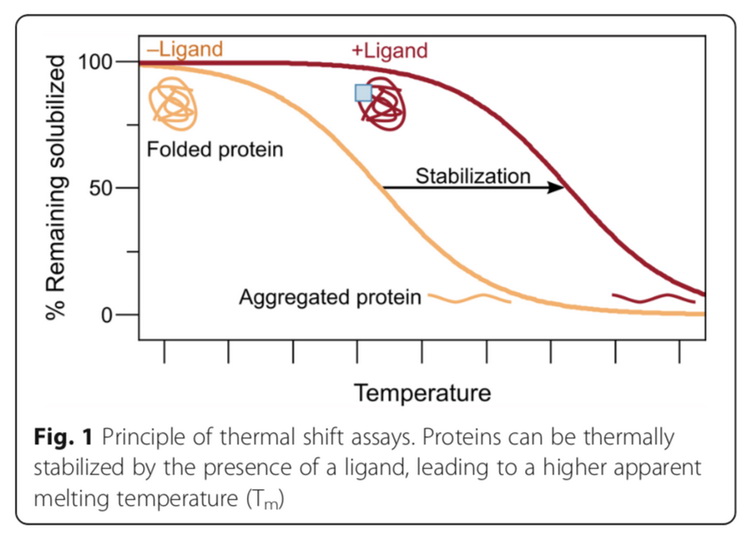
图9 热蛋白质组学的原理(André Mateus, Tomi A. Määttä and Mikhail M. Savitski. Proteome Science, 2017, 15:13)
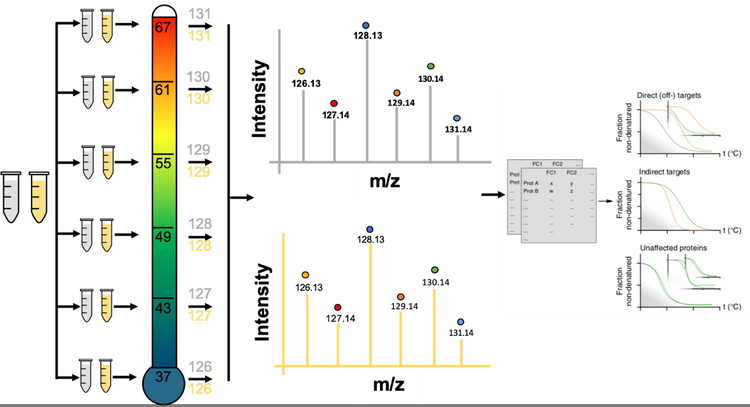
图10 鉴定活性分子靶标蛋白的热蛋白质组学实验步骤
我们还在不断发展其它质谱和蛋白质组学的新方法,如同学们有质谱方面的特殊需求,或者想了解更多的信息,欢迎各位老师同学和我们交流!联系电话:010-62789417转211
送样地址:清华大学医学科学楼B-1014房间(2020年春节后送样地址变更为生物医学馆地下U5层)。
<em font-size:16px;text-align:right;text-indent:32px;white-space:normal;background-color:#ffffff;"="" style="box-sizing:border-box;outline:0px;text-size-adjust:none;-webkit-tap-highlight-color:rgba(0, 0, 0, 0);margin:0px;padding:0px;color:#808080">本文章版权归清华大学蛋白质研究技术中心蛋白质化学与组学平台所有
